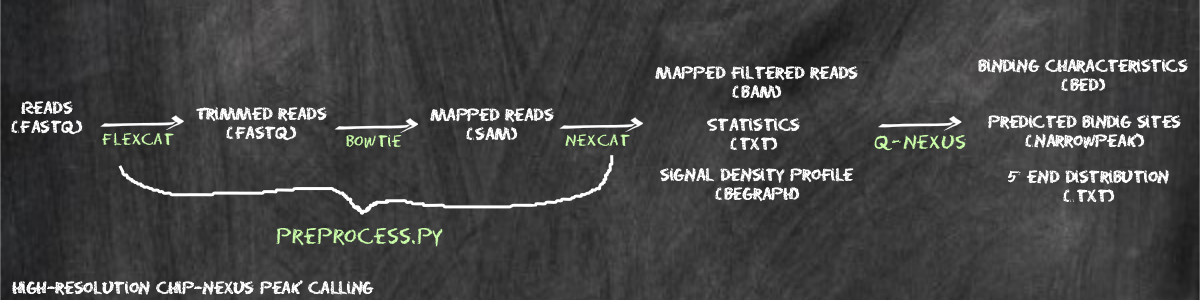
Q-nexus is a peak caller designed for the analysis of ChIP-nexus data. ChIP-nexus (chromatin immunoprecipitation experiments with nucleotide resolution through exonuclease, unique barcode and single ligation), is an extension of ChIP-seq and ChIP-exo that utilizes an efficient DNA self-circularization step during library preparation. This allows protein-DNA binding sites to be characterized to base-pair resolution. Q-nexus additionally can be used to analyze ChIP-exo data. This tutorial explains how to install and use Q-nexus, and presents a complete example using the a dataset in which the transcription factor dorsal was investigated in Drosophila melanogaster.
The entire Q-nexus pipeline requires three programs.
This Manual is subdivided into the following sections:
In the first step, we process the raw FASTQ file generated by the ChIP-nexus experiment in order to perform adapter clipping and simultaneously identify the Q-nexus random barcodes. We note that the random barcode is followed in the 3' direction by a four nucleotide fixed barcode (See Figure 1A of the Q-nexus manuscript). Reads that do not have this fixed barcode are likely to be artefacts and are removed by flexcat. The fixed barcode and the random barcode are removed from the sequence but the random barcode is added to the ID line of the output FASTQ file.
ChIP-exo data do not have a fixed or random barcode. Preprocessing therefore involves only adaptor trimming.
As an optional step for both ChIP-nexus and ChIP-exo data, quality trimming can be performed.
The following options are used for this application| Option | Description |
|---|---|
| -tl 5 | Trim 5 bases from the 5" end of each read, this removes the random barcode from the read sequence |
| -tt | Store trimmed sequences in the corresponding id, this is neccessary to process the trimmed random barcode at a later stage |
| -t | Tag sequences with removed adapters |
| -ml 0 | Set the final minimum length of reads that will be accepted, default is 0 |
| -er 0.2 | Error rate for adapter matching, default is 0.2 |
| -ol 4 | Minimum required overlap for adapter matching, default is 4 |
| -app | Allow 1 base error for fixed barcode matching |
| -tnum | Number of threads, set according to tnum script parameter |
| -ss | Display processing speed in reads per second |
| -fr [N] | Process only first N reads, default is all reads |
flexcat requires two input files that specify the adapter sequences to be trimmed as well as the fixed barcode. The adapter sequences can be specified in the file adapters.fa. For example, the following file can be used for the adaptor in the original Q-nexus protocol ( which is located in the Q/data/ directory. This file could for example look like this
>adapter1
AGATCGGAAGAGCACACGTCTGGATCCACGACGCTCTTCC
>adapter2
GATCGGAAGAGCACACGTCTGGATCCACGACGCTCTTCC
>adapter3
ATCGGAAGAGCACACGTCTGGATCCACGACGCTCTTCC
>adapter4
TCGGAAGAGCACACGTCTGGATCCACGACGCTCTTCC
For single-end reads, adapter*.1 is omitted. The adapters in this example file are chosen to match ChiP-nexus protocol. If you use another another protocol, for example ChiP-Exo, this file needs to be modified accordingly.
ChiP-nexus reads have in contrast to ChiP-Exo reads two barcodes at the 5"-end of each read. The first barcode is a random barcode of length 5, the second barcode immediately follows the first barcode and is a fixed barcode of length 4. The fixed barcode is used to identify valid ChiP-nexus reads. Therefore Reads at this processing step will be filtered for the fixed barcode, and only those who match the fixed barcode in Q/data/barcodes.fa with an allowed matching error of 1 base will be used for further processing. For the standard ChiP-nexus protocol, the barcodes.fa file looks like this
>matched_barcode
CTGA Mapping is done using Bowtie 1.1.2. Although there is already Bowtie 2 available, the authors of Bowtie say, that Bowtie 1.1.2 is better suitable for short reads. The following options are used for bowtie
| Option | Description |
|---|---|
| -k 1 | report up to 1 valid alignment per read |
| -m 1 | only map reads that are uniquely mappable |
| --chunkmbs 512 | each thread can use 512 MBs memory |
| --strata --best | See Bowtie manual for detailed |
| -S | Output file format is SAM |
| -p | Number of threads, set according to tnum script parameter |
| -v 2 | Set verbosity level to 2, however this output will only be displayed, if the script is executed with --verbose |
The mapped reads need to be further processed, because for ChiP-nexus we want to remove reads that are mapped to the same position and have the same random barcode. This enabled us to distinguish PCR artifact duplicates from real duplicates. For this preprocessing stage, the binary nexus-pre is used with the following settings
| Option | Description |
|---|---|
| -fc [REGEX] | regular expression to remove chromosomes from the calcuation of the qfragment-length-distribution |
We provide a script called preprocess.py, that enabled you to run all the above mentioned preprocessing steps with one command. We recommend the following directory structure.
| Directory | Description |
|---|---|
| data | a directory where all the input and output data of the preprocessing resides |
| data/fastq | raw reads |
| data/genomes | reference genomes |
| data/preprocessed | directory where all the output data of the preprocessing goes |
Now you can start the preprocessing pipeline be executing this command
~/Q/scripts/preprocess.py ~/data/fastq/input_reads_ID01.fastq ~/data/genome/ref_genome.fa --clean --output_dir ~/data/preprocessed/ID01 --verbose --overwrite
CTGA The following table explains the options that can be used with this script
| Option | Description |
|---|---|
| [INPUT FILE] | path the the input file with raw reads (fastq or fastq.gz) |
| [GENOME FILE] | path to the reference genome file (fasta or fasta.gz) |
| --exo | don't do barcode trimming (use this option if you process ChiP-Exo reads) |
| --clean | delete intermediate files, such as the mapping output generated by bowtie |
| --overwrite | overwrite existing output files, if this option is not used, preprocessing steps will be skipped if the corresponding output file already exists |
| --verbose | display output of the different preprocessing stages on screen |
| --bowtie_location [PATH] | specify the path to the bowtie executable, this is neccessary if bowtie is not in the path and for all windows installations |
| --num_threads [N] | use N threads, default is 4 |
| --output_dir [PATH] | directory where the output files will be created, default is the current working directory |
| --filter_chromosomes [REGEX] | regular expression to remove chromosomes from the calcuation of the qfragment-length-distribution |
The preprocessing pipeline generates a file which is called [INPUT_PREFIX]_duplication_rate.txt. This file containts information about how many reads with a specific coverage rate exist. We also supply and R-script that can generate the following graph out of this txt file.
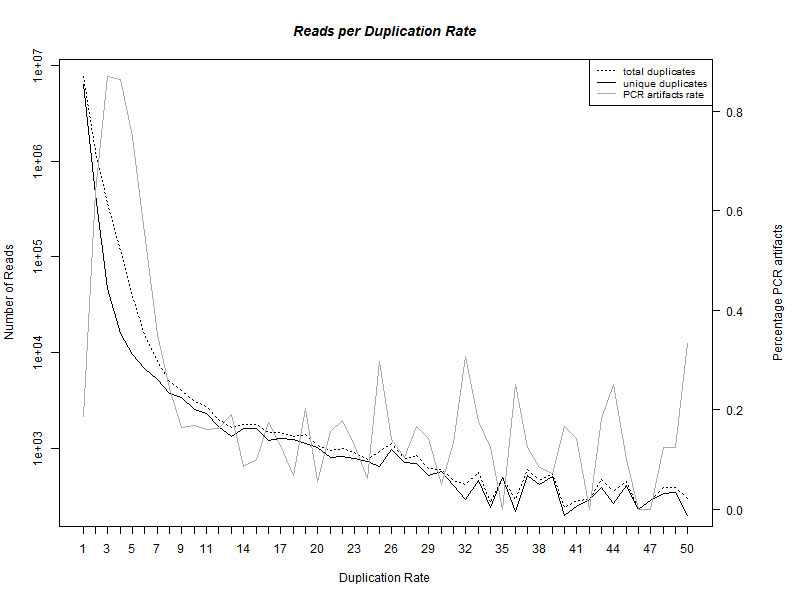
On the x-axis of this graph is the coverage rate, the left y-axis show the number of reads. The right y-axis show the percentage of non unique duplet reads out of all duplet reads. The discrimination of unique and non unique duplicates is for the first time possible, because of the random barcodes used by the ChiP-nexus protocol. The preprocessing output can be further analyzed using the tool FastQC.
The preprocessing pipeline also generates a file with statistical information about the preprocessed data. It could look like this
Total reads 17102794
Filtered reads (-fc Option) 0
Mapped reads 9616348
Non mappable reads 2082353
Non uniquely mappable reads 5404093
After barcode filtering 6891129 (-2725219)
PCR duplication rate 0.283394
Total duplet reads 1274581
Estimated fragment Length 18Using the switch --nexus-mode will cause Q to use parameter settings optimized for ChIP-nexus data. The usage is described in the Q-nexus tutorial. For a comprehensive list of all parameters type:
Q --help In --nexus-mode Q will automatically derive the width of the protected region using the distribution of qfrag lengths with pseudo-control. Then peak calling will be performed using the estimated width.
Q -nexus-mode -t IN.bam -o OUT_PREFIX Peak calling can be skipped using the --binding-characteristics-only option.
Q --nexus-mode --binding-characteristics-only -t IN.bam -o OUT_PREFIX The estimation of the protected region width can be skipped by specifying the width using the -l option.
Q --nexus-mode -l 12 -t IN.bam -o OUT_PREFIX The “-w” option causes Q to output all qfrags for a given chromosome in BED format.
Q --nexus-mode -w chr2L -t IN.bam -o chr2L In order to view the qfrags coverage profile in IGV the BED file has to be sorted and converted into tdf format using igvtools, as follows:
igvtools sort chr2L-qfrags.bed chr2L-qfrags-sorted.bedigvtools count -z 1 -w 1 --includeDuplicates chr2L-qfrags-sorted.bed chr2L-qfrags-sorted.tdf dm3.chrom.sizes